Apert Sendromu
+Apert sendromu, kafatasının, yüzün, ellerin ve ayakların malformasyonları ile karakterize bir konjenital bozukluk olan bir akrosefalosindaktili formudur. Çene ve mandibulanın öncüsü olan birinci branşiyal (veya faringeal) kemeri etkileyen bir dalsal ark sendromu olarak sınıflandırılır. Fetal gelişimde branşiyal arkların gelişimindeki bozukluklar kalıcı ve yaygın etkiler yaratır.
+1906'da Fransız bir hekim olan Eugène Apert, benzer özellikleri ve özellikleri paylaşan dokuz kişiyi tanımladı. Dilbilimsel olarak, "akrosefalosindaktili" teriminde, akro, sendromda yaygın olan "sivri uçlu" baş anlamına gelen "tepe" anlamına gelen Yunanca'dır; yine Yunancadan olan cephalo, "kafa" anlamına gelen birleşik bir formdur; sindaktili olarak parmakların ve ayak parmaklarının dokumasını ifade eder.
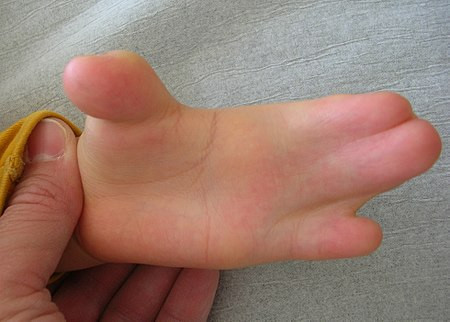
Embriyolojide, ellerde ve ayaklarda, selektif hücre ölümü veya apoptoz adı verilen ve parmakların ayrılmasına neden olan bir süreçte ölen seçici hücreler bulunur. Akrosefalosindaktili durumunda, seçici hücre ölümü meydana gelmez ve cilt ve nadiren de kemik, parmaklar ve ayak parmakları arasında kaynaşır.
+Crouzon sendromu ve Pfeiffer sendromuna benzer şekilde kraniyal kemikler de etkilenir. Kraniosinostoz, fetal kafatası ve yüz kemiklerinin uteroda çok erken kaynaşması ve normal kemik büyümesini bozması durumunda ortaya çıkar. Farklı sütürlerin füzyonu, kafatasında farklı büyüme modellerine yol açar. Örnekler şunları içerir: trigonosefali (metopik sütürün füzyonu), brakisefali (iki taraflı koronal sütür ve lambdoid sütürün füzyonu), dolikosefali (sagittal sütürün füzyonu), plajiyosefali (koronal ve lambdoidal sütürlerin tek taraflı olarak füzyonu) ve oksiefali füzyonu koronal ve lambdoid sütürler).
+Popülasyondaki sendrom insidansına ilişkin bulgular, 200.000'de 1 doğum kadar düşük tahminlerle ve daha eski çalışmalar tarafından ortalama olarak 160.000 olarak verilmiştir. Ancak, 1997'de California Doğum Kusurları İzleme Programı tarafından yapılan bir çalışmada, yaklaşık 2,5 milyon canlı doğumdan 80.645'te 1 insidans oranı bulundu. 2002 yılında Craniofacial Center, North Texas Hospital For Children tarafından yürütülen bir başka çalışmada, 65.000 canlı doğumda yaklaşık 1 oranında daha yüksek bir insidans bulundu.
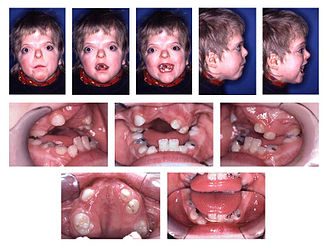
+Kraniyal malformasyonlar, akrosefalosindaktiliğin en belirgin etkileridir. Kraniosinostoz, çocuğun beyni büyümeye ve genişlemeye devam etmesine rağmen kraniyal sütürlerin çok erken kapanmasıyla oluşur. Brakisefali, koronal sütürlerin erken kapandığı, kafatasının öne veya arkaya doğru genişlemesini önlediği ve beynin kafatasını yanlara ve yukarıya doğru genişletmesine neden olan yaygın büyüme modelidir. Bu, başka bir ortak özellik ile sonuçlanır: yüksek, belirgin bir alın ve kafatasının arkası düzdür. Koronal sütürlerin erken kapanması nedeniyle, artmış kafa basıncı gelişerek zihinsel yetersizliğe yol açabilir. Orta yüz kemiklerindeki yetersiz büyümenin bir sonucu olarak düz veya içbükey bir yüz gelişebilir ve bu da psödomandibular prognatizm olarak bilinen bir duruma yol açar. Akrosefalosindaktilinin diğer özellikleri sığ kemikli yörüngeleri ve geniş aralıklı gözleri içerebilir. Düşük ayarlanmış kulaklar aynı zamanda branşiyal ark sendromlarının tipik bir özelliğidir.
+Akrosefalosindaktili ile ilgili ortak özellikler, yüksek kemerli bir damak, psödomandibuler prognatizm (mandibular prognatizm olarak ortaya çıkan), dar bir damak ve dişlerin çapraşıklığıdır.
+Teşhis tipik olarak görünen fiziksel özelliklerle yapılır ve kafatası röntgeni veya kafa BT muayenesi ile yardımcı olabilir. Moleküler genetik testler tanıyı doğrulayabilir.
KAYNAK: https://en.wikipedia.org/wiki/Apert_syndrome